Related Publications
Parallel Protein Folding with STAPL, Shawna Thomas, Gabriel Tanase, Lucia K. Dale, Jose E. Moreira, Lawrence Rauchwerger, Nancy M. Amato, Concurrency and Computation: Practice and Experience, Vol: 17, Issue: 14, pp. 1643-1656, Dec 2005. DOI: https://doi.org/10.1002/cpe.950
Keywords: Parallel Planning, Protein Folding, STAPL
Links : [Published]
BibTex
@article{https://doi.org/10.1002/cpe.950,
author = {Thomas, Shawna and Tanase, Gabriel and Dale, Lucia K. and Moreira, Jose M. and Rauchwerger, Lawrence and Amato, Nancy M.},
title = {Parallel protein folding with STAPL},
journal = {Concurrency and Computation: Practice and Experience},
volume = {17},
number = {14},
pages = {1643-1656},
keywords = {protein folding, motion planning, parallel libraries, C++},
doi = {https://doi.org/10.1002/cpe.950},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/cpe.950},
eprint = {https://onlinelibrary.wiley.com/doi/pdf/10.1002/cpe.950},
abstract = {Abstract The protein-folding problem is a study of how a protein dynamically folds to its so-called native state—an energetically stable, three-dimensional conformation. Understanding this process is of great practical importance since some devastating diseases such as Alzheimer's and bovine spongiform encephalopathy (Mad Cow) are associated with the misfolding of proteins. We have developed a new computational technique for studying protein folding that is based on probabilistic roadmap methods for motion planning. Our technique yields an approximate map of a protein's potential energy landscape that contains thousands of feasible folding pathways. We have validated our method against known experimental results. Other simulation techniques, such as molecular dynamics or Monte Carlo methods, require many orders of magnitude more time to produce a single, partial trajectory. In this paper we report on our experiences parallelizing our method using STAPL (Standard Template Adaptive Parallel Library) that is being developed in the Parasol Lab at Texas A\&M. An efficient parallel version will enable us to study larger proteins with increased accuracy. We demonstrate how STAPL enables portable efficiency across multiple platforms, ranging from small Linux clusters to massively parallel machines such as IBM's BlueGene/L, without user code modification. Copyright © 2005 John Wiley \& Sons, Ltd.},
year = {2005}
}
Abstract
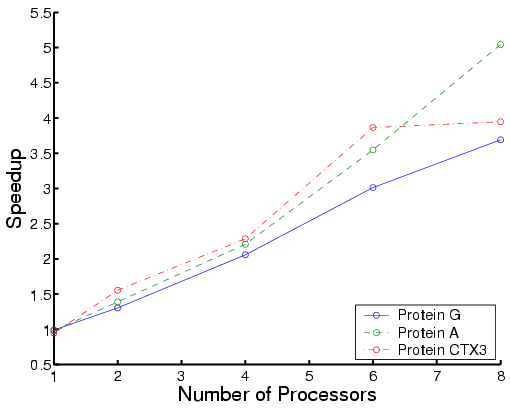
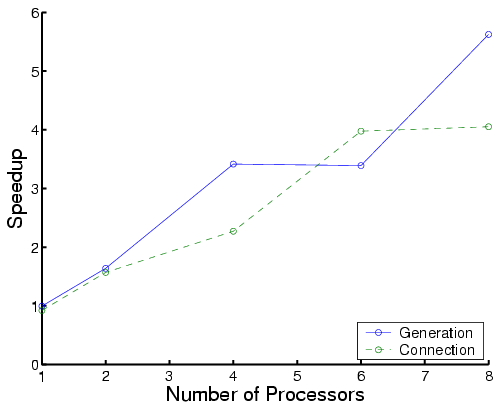
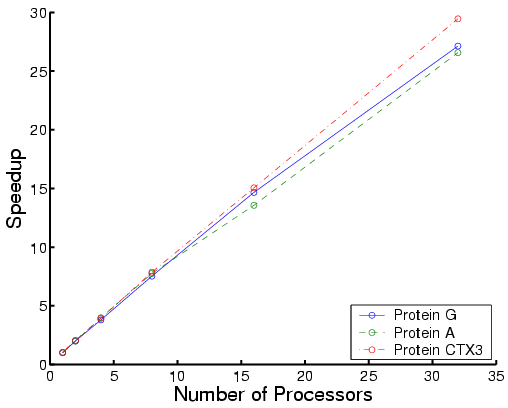
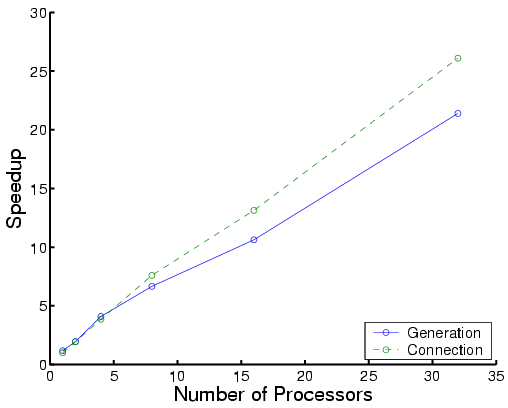
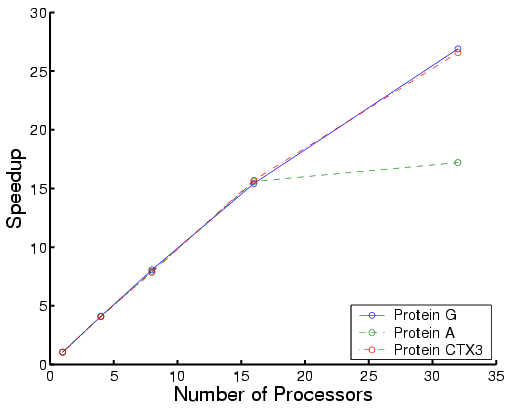
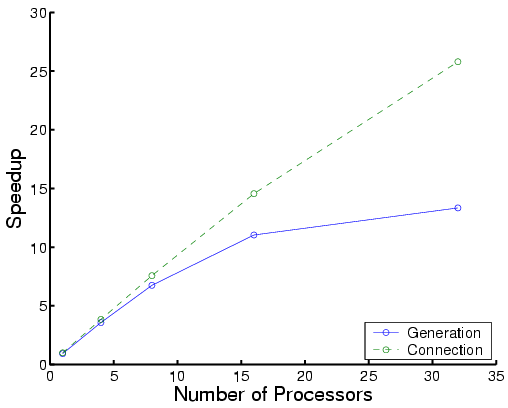
The protein-folding problem is a study of how a protein dynamically folds to its so-called native state - an energetically stable, three-dimensional conformation. Understanding this process is of great practical importance since some devastating diseases such as Alzheimer's and bovine spongiform encephalopathy (Mad Cow) are associated with the misfolding of proteins. We have developed a new computational technique for studying protein folding that is based on probabilistic roadmap methods for motion planning. Our technique yields an approximate map of a protein's potential energy landscape that contains thousands of feasible folding pathways. We have validated our method against known experimental results. Other simulation techniques, such as molecular dynamics or Monte Carlo methods, require many orders of magnitude more time to produce a single, partial trajectory. In this paper we report on our experiences parallelizing our method using STAPL (Standard Template Adaptive Parallel Library) that is being developed in the Parasol Lab at Texas A&M. An efficient parallel version will enable us to study larger proteins with increased accuracy. We demonstrate how STAPL enables portable efficiency across multiple platforms, ranging from small Linux clusters to massively parallel machines such as IBM's BlueGene/L, without user code modification.
Parallel Protein Folding with STAPL, Shawna Thomas, Nancy M. Amato, 18th International Parallel and Distributed Processing Symposium (IPDPS), pp. 189-, Santa Fe, NM, USA, Apr 2004. DOI: 10.1109/IPDPS.2004.1303204
Keywords: Parallel Planning, Protein Folding, STAPL
Links : [Published]
BibTex
@INPROCEEDINGS{1303204,
author={S. {Thomas} and N. M. {Amato}},
booktitle={18th International Parallel and Distributed Processing Symposium, 2004. Proceedings.},
title={Parallel protein folding with STAPL},
year={2004},
volume={},
number={},
pages={189-},
doi={10.1109/IPDPS.2004.1303204}}
Abstract
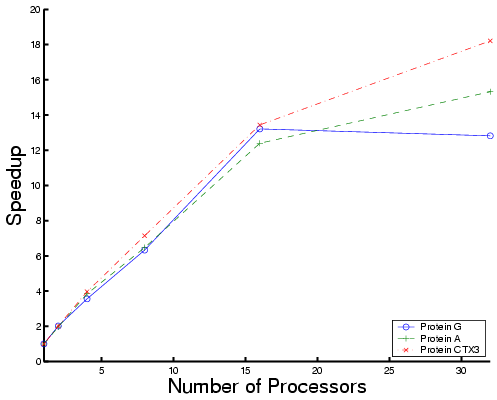
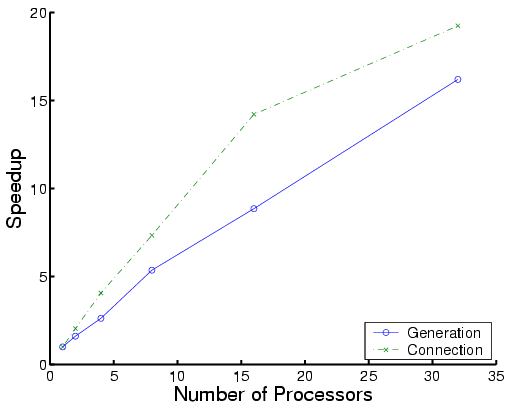
Summary form only given. The protein folding problem is to study how a protein dynamically folds to its so-called native state - an energetically stable, three-dimensional configuration. Understanding this process is of great practical importance since some devastating diseases such as Alzheimer\'s and bovine spongiform encephalopathy (Mad Cow) are associated with the misfolding of proteins. In our group, we have developed a new computational technique for studying protein folding that is based on probabilistic roadmap methods for motion planning. Our technique yields an approximate map of a protein\'s potential energy landscape that contains thousands of feasible folding pathways. We have validated our method against known experimental results. Other simulation techniques, such as molecular dynamics or Monte Carlo methods, require many orders of magnitude more time to produce a single, partial, trajectory. We report on our experiences parallelizing our method using STAPL (the standard template adaptive parallel library), that is being developed in the Parasol Lab at Texas A&M. An efficient parallel version enables us to study larger proteins with increased accuracy. We demonstrate how STAPL enables portable efficiency across multiple platforms without user code modification. We show performance gains on two systems: a dedicated Linux cluster and an extremely heterogeneous multiuser Linux cluster.