Related Publications
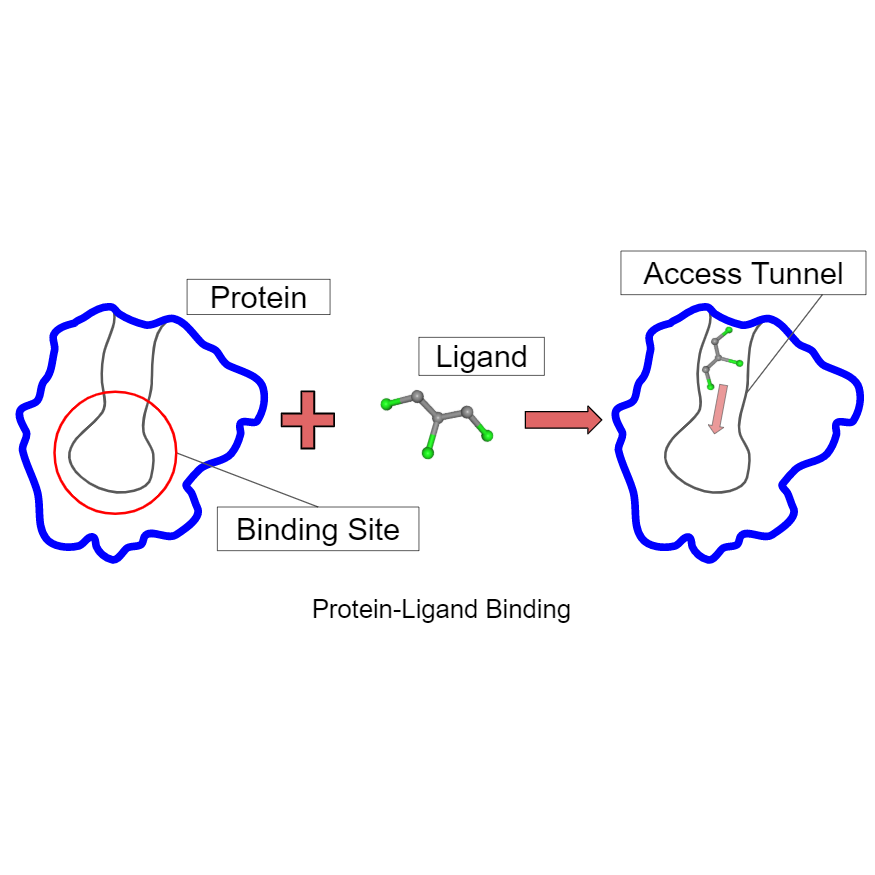
Using Guided Motion Planning to Study Binding Site Accessibility, Diane Uwacu, Abigail Ren, Shawna Thomas, Nancy M. Amato, Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Issue: 109, (Virtual) New York, USA, Sep 2020. DOI: 10.1145/3388440.3414707
Keywords: Computational Biology, Ligand Binding, Motion Planning
Links : [Published] [Manuscript]
BibTex
@inbook{10.1145/3388440.3414707,
author = {Uwacu, Diane and Ren, Abigail and Thomas, Shawna and Amato, Nancy M.},
title = {Using Guided Motion Planning to Study Binding Site Accessibility},
year = {2020},
isbn = {9781450379649},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3388440.3414707},
abstract = {Computational methods are commonly used to predict protein-ligand interactions. These methods typically search for regions with favorable energy that geometrically fit the ligand, and then rank them as potential binding sites. While this general strategy can provide good predictions in some cases, it does not do well when the binding site is not accessible to the ligand. In addition, recent research has shown that in some cases protein access tunnels play a major role in the activity and stability of the protein's binding interactions. Hence, to fully understand the binding behavior of such proteins, it is imperative to identify and study their access tunnels. In this work, we present a motion planning algorithm that scores protein binding site accessibility for a particular ligand. This method can be used to screen ligand candidates for a protein by eliminating those that cannot access the binding site. This method was tested on two case studies to analyze effects of modifying a protein's access tunnels to increase activity and/or stability as well as study how a ligand inhibitor blocks access to the protein binding site.},
booktitle = {Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics},
articleno = {109},
numpages = {10}
}
Abstract
Computational methods are commonly used to predict protein-ligand interactions. These methods typically search for regions with favorable energy that geometrically fit the ligand, and then rank them as potential binding sites. While this general strategy can provide good predictions in some cases, it does not do well when the binding site is not accessible to the ligand. In addition, recent research has shown that in some cases protein access tunnels play a major role in the activity and stability of the protein's binding interactions. Hence, to fully understand the binding behavior of such proteins, it is imperative to identify and study their access tunnels. In this work, we present a motion planning algorithm that scores protein binding site accessibility for a particular ligand. This method can be used to screen ligand candidates for a protein by eliminating those that cannot access the binding site. This method was tested on two case studies to analyze effects of modifying a protein's access tunnels to increase activity and/or stability as well as study how a ligand inhibitor blocks access to the protein binding site.
Using Motion Planning to Evaluate Protein Binding Site Accessibility, Diane Uwacu, Everett Yang, Shawna Thomas, Nancy M. Amato, Technical Report, TR18-001, Parasol Laboratory, Department of Computer Science, Texas A&M University, College Station TX 77848, USA, Jul 2018.
Keywords: Ligand Binding, Motion Planning, Workspace Topology
Links : [Manuscript]
BibTex
N/A
Abstract
Despite many efforts and considerable breakthroughs in ligand binding prediction, the best predictors still produce many false positives and a reliable,
fully automated prediction framework has yet to be developed. Binding site accessibility is an important feature ignored by methods that classify binding based solely on the energetic or geometric properties of the final bound protein-ligand complex. To evaluate this necessity, we transform the ligand accessibility problem into a robot motion planning problem where the ligand is modeled as a flexible agent whose task is to travel from outside the protein to its binding site. We use Rapidly-exploring Random Graphs coupled with Mean Curve workspace skeletons to quickly and thoroughly explore a protein environment in order to produce valid paths for ligand motion. Path weights reflect the influences of intermolecular forces on the given ligand. Low weight paths are extracted and analyzed for characteristics of accessibility. In this paper, we show that our algorithm provides a mechanism to evaluate binding site accessibility for a ligand.
Adaptive Local Learning in Sampling Based Motion Planning for Protein Folding, Chinwe Ekenna and Shawna Thomas and Nancy M. Amato, BMC Syst Biol, Special Issue from the 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Vol: 10, Issue: 49, pp. 165-179, Aug 2016. DOI: 10.1186/s12918-016-0297-9
Keywords: Computational Biology, Protein Folding
Links : [Published]
BibTex
@article{ekenna-bmcsb-2016
, author = "Chinwe Ekenna and Shawna Thomas and Nancy M. Amato"
, title = "Adaptive Local Learning in Sampling Based Motion Planning for Protein Folding"
, journal = "BMC Syst Biol, Special Issue from IEEE International Conference on Bioinformatics and Biomedicine (BIBM)"
, volume = "10"
, number = "49"
, pages = "165--179"
, year = "2016"
, month = "August"
, doi = "10.1186/s12918-016-0297-9"
}
Abstract
Background
Simulating protein folding motions is an important problem in computational biology. Motion planning algorithms, such as Probabilistic Roadmap Methods, have been successful in modeling the folding landscape. Probabilistic Roadmap Methods and variants contain several phases (i.e., sampling, connection, and path extraction). Most of the time is spent in the connection phase and selecting which variant to employ is a difficult task. Global machine learning has been applied to the connection phase but is inefficient in situations with varying topology, such as those typical of folding landscapes.
Results
We develop a local learning algorithm that exploits the past performance of methods within the neighborhood of the current connection attempts as a basis for learning. It is sensitive not only to different types of landscapes but also to differing regions in the landscape itself, removing the need to explicitly partition the landscape. We perform experiments on 23 proteins of varying secondary structure makeup with 52ÃÂÃÂÃÂâÃÂÃÂÃÂÃÂÃÂÃÂÃÂÃÂ114 residues. We compare the success rate when using our methods and other methods. We demonstrate a clear need for learning (i.e., only learning methods were able to validate against all available experimental data) and show that local learning is superior to global learning producing, in many cases, significantly higher quality results than the other methods.
Conclusions
We present an algorithm that uses local learning to select appropriate connection methods in the context of roadmap construction for protein folding. Our method removes the burden of deciding which method to use, leverages the strengths of the individual input methods, and it is extendable to include other future connection methods.
Decoy Database Improvement for Protein Folding, Hsin-Yi (Cindy) Yeh, Aaron Lindsey, Chih-Peng Wu, Shawna Thomas, and Nancy M. Amato, Journal of Computational Biology, Vol: 22, Issue: 9, pp. 823-836, Sep 2015. DOI: 10.1089/cmb.2015.0116
Keywords: Computational Biology, Protein Folding
Links : [Published]
BibTex
@article{yeh-jcb-2015
, author = "Hsin-Yi Yeh and Aaron Lindsey and Chih-Peng Wu and Shawna Thomas and Nancy M. Amato"
, title ="Decoy Database Improvement for Protein Folding"
, journal = "Journal of Computational Biology"
, volume = "22"
, number = "9"
, year = "2015"
, pages = "823-836"
, doi = "10.1089/cmb.2015.0116"
}
Abstract
Predicting protein structures and simulating protein folding are two of the most important problems in computational biology today. Simulation methods rely on a scoring function to distinguish the native structure (the most energetically stable) from non-native structures. Decoy databases are collections of non-native structures used to test and verify these functions.
We present a method to evaluate and improve the quality of decoy databases by adding novel structures and removing redundant structures. We test our approach on 20 different decoy databases of varying size and type and show significant improvement across a variety of metrics. We also test our improved databases on two popular modern scoring functions and show that for most cases they contain a greater or equal number of native-like structures than the original databases, thereby producing a more rigorous database for testing scoring functions.
Rigidity Analysis for Protein Motion and Folding Core Identification, Shawna Thomas, Lydia Tapia, Chinwe Ekenna, Hsin-Yi (Cindy) Yeh, Nancy M. Amato, Association for the Advancement of Artificial Intelligence (AAAI) Workshop, Vol: WS-13-06, pp. 38-43, Bellevue, Washington, Jul 2013.
Keywords: Computational Biology, Protein Folding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@inproceedings{thomas-aaaiW-2013
, author = "Shawna Thomas and Lydia Tapia and Chinwe Ekenna and Hsin-Yi (Cindy) Yeh and Nancy M. Amato "
, title = "Rigidity Analysis for Protein Motion and Folding Core Identification"
, booktitle = "AAAI Workshop on Artificial Intelligence and Robotics Methods in Computational Biology"
, volume = "WS-13-06"
, month = "July"
, address = "Bellevue, Washington, USA"
, pages = "38-43"
, year = "2013"
}
Abstract
Protein folding plays an essential role in protein function and stability. Despite the explosion in our knowledge of structural and functional data, our understanding of protein folding is still very limited. In addition, methods such as folding core identification are gaining importance with the increased desire to engineer proteins with particular functions and efficiencies. However, defining the folding core can be challenging for both experiment and simulation. In this work, we use rigidity analysis to effectively sample and model the protein's energy landscape and identify the folding core. Our results show that rigidity analysis improves the accuracy of our approximate landscape models and produces landscape models that capture the subtle folding differences between protein G and its mutants, NuG1 and NuG2. We then validate our folding core identification against known experimental data and compare to other simulation tools. In addition to correlating well with experiment, our method can suggest other components of structure that have not been identified as part of the core because they were not previously measured experimentally.
A Multi-Directional Rapidly Exploring Random Graph (mRRG) for Protein Folding, Shuvra Nath, Shawna Thomas, Chinwe Ekenna, Nancy M. Amato, Proc. ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB), pp. 44-51, Orlando, FL, USA, Oct 2012. DOI: 10.1145/2382936.2382942
Keywords: Computational Biology, Protein Folding
Links : [Published]
BibTex
@inproceedings{nath-bcb-2012
, author = "Shuvra Nath and Shawna Thomas and Chinwe Ekenna and Nancy M. Amato"
, title = "A Multi-Directional Rapidly Exploring Random Graph (mRRG) for Protein Folding"
, booktitle = "ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB)"
, pages = "44-51"
, location ="Orlando, FL, USA"
, month = "October"
, year = "2012"
, doi = "10.1145/2382936.2382942"
}
Abstract
Modeling large-scale protein motions, such as those involved in folding and binding interactions, is crucial to better understanding not only how proteins move and interact with other molecules but also how proteins misfold, thus causing many devastating diseases. Robotic motion planning algorithms, such as Rapidly Exploring Random Trees (RRTs), have been successful in simulating protein folding pathways. Here, we propose a new multi-directional Rapidly Exploring Random Graph (mRRG) specifically tailored for proteins.
Unlike traditional RRGs which only expand a parent conformation in a single direction, our strategy expands the parent conformation in multiple directions to generate new samples. Resulting samples are connected to the parent conformation and its nearest neighbors. By leveraging multiple directions, mRRG can model the protein motion landscape with reduced computational time compared to several other robotics-based methods for small to moderate-sized proteins. Our results on several proteins agree with experimental hydrogen out-exchange, pulse-labeling, and æ-value analysis. We also show that mRRG covers the conformation space better as compared to the other computation methods.
A Motion Planning Approach to Studying Molecular Motions, Lydia Tapia, Shawna Thomas, Nancy M. Amato, Communications in Information and Systems, Vol: 10, Issue: 1, pp. 53-68, Jan 2010. DOI: 10.4310/cis.2010.v10.n1.a4
Keywords: Computational Biology, Motion Planning, Protein Folding
Links : [Published]
BibTex
@article{tapia-cis-2010-j
, author = "Lydia Tapia and Shawna Thomas and Nancy M. Amato"
, title = "A Motion Planning Approach to Studying Molecular Motions"
, journal = "Communications in Information and Systems"
, volume = "10"
, issue = "1"
, year = "2010"
, pages = "52-68"
, doi = "10.4310/CIS.2010.v10.n1.a4"
, note = "Special Issue in honor of Michael Waterman. Open Access. https://projecteuclid.org/euclid.cis/1268143373"
}
Abstract
While structurally very different, protein and RNA molecules share an important attribute. The motions they undergo are strongly related to the function they perform. For example, many diseases such as Mad Cow disease or AlzheimerâÃÂÃÂs disease are associated with protein misfolding and aggregation. Similarly, RNA folding velocity may regulate the plasmid copy number, and RNA folding kinetics can regulate gene expression at the translational level. Knowledge of the stability, folding, kinetics and detailed mechanics of the folding process may help provide insight into how proteins and RNAs fold. In this paper, we present an overview of our work with a computational method we have adapted from robotic motion planning to study molecular motions. We have validated against experimental data and have demonstrated that our method can capture biological results such as stochastic folding pathways, population kinetics of various conformations, and relative folding rates. Thus, our method provides both a detailed view (e.g., individual pathways) and a global view (e.g., population kinetics, relative folding rates, and reaction coordinates) of energy landscapes of both proteins and RNAs. We have validated these techniques by showing that we observe the same relative folding rates as shown in experiments for structurally similar protein molecules that exhibit different folding behaviors. Our analysis has also been able to predict the same relative gene expression rate for wild-type MS2 phage RNA and three of its mutants.
Kinetics Analysis Methods for Approximate Folding Landscapes , Lydia Tapia, Xinyu Tang, Shawna Thomas, Nancy M. Amato, Bioinformatics, Vol: Volume 23, Issue: Issue 13, pp. i539- i548, Jul 2007. DOI: 10.1093/bioinformatics/btm199
Keywords: Protein Folding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@article{tapia2007kinetics,
title={Kinetics analysis methods for approximate folding landscapes},
author={Tapia, Lydia and Tang, Xinyu and Thomas, Shawna and Amato, Nancy M},
journal={Bioinformatics},
volume={23},
number={13},
pages={i539--i548},
year={2007},
publisher={Oxford University Press}
}
Abstract
Motivation:
Protein motions play an essential role in many
biochemical processes. Lab studies often quantify these motions
in terms of their kinetics such as the speed at which a protein folds or
the population of certain interesting states like the native state.
Kinetic metrics give quantifiable measurements of the folding
process that can be compared across a group of proteins such as
a wild-type protein and its mutants.
Results:
We present two new techniques, map-based master
equation solution and map-based Monte Carlo simulation, to study
protein kinetics through folding rates and population kinetics from
approximate folding landscapes, models called maps. From these
two new techniques, interesting metrics that describe the folding
process, such as reaction coordinates, can also be studied. In this
article we focus on two metrics, formation of helices and structure
formation around tryptophan residues. These two metrics are
often studied in the lab through circular dichroism (CD) spectra
analysis and tryptophan fluorescence experiments, respectively.
The approximated landscape models we use here are the maps of
protein conformations and their associated transitions that we have
presented and validated previously.
In contrast to other methods such as the traditional master
equation and Monte Carlo simulation, our techniques are both fast
and can easily be computed for full-length detailed protein models.
We validate our map-based kinetics techniques by comparing
folding rates to known experimental results. We also look in depth
at the population kinetics, helix formation and structure near
tryptophan residues for a variety of proteins.
Tools for Simulating and Analyzing RNA Folding Kinetics, Xinyu Tang, Shawna Thomas, Lydia Tapia, Nancy M. Amato, In. Proc. International Conference on Research in Computational Molecular Biology, Oaklanda, California, USA, Apr 2007. DOI: 10.1007/978-3-540-71681-5_19
Keywords: Computational Biology, RNA, Sampling-Based Motion Planning
Links : [Published]
BibTex
@InProceedings{10.1007/978-3-540-71681-5_19,
author="Tang, Xinyu
and Thomas, Shawna
and Tapia, Lydia
and Amato, Nancy M.",
editor="Speed, Terry
and Huang, Haiyan",
title="Tools for Simulating and Analyzing RNA Folding Kinetics",
booktitle="Research in Computational Molecular Biology",
year="2007",
publisher="Springer Berlin Heidelberg",
address="Berlin, Heidelberg",
pages="268--282",
isbn="978-3-540-71681-5"
}
Abstract
It has recently been found that some RNA functions are determined by the actual folding kinetics and not just the RNAâÃÂÃÂs nucleotide sequence or its native structure. We present new computational tools for simulating and analyzing RNA folding kinetic metrics such as population kinetics, folding rates, and the folding of particular subsequences. Our method first builds an approximate representation (called a map) of the RNAâÃÂÃÂs folding energy landscape, and then uses specialized analysis techniques to extract folding kinetics from the map. We provide a new sampling strategy called Probabilistic Boltzmann Sampling (PBS) that enables us to approximate the folding landscape with much smaller maps, typically by several orders of magnitude. We also describe a new analysis technique, Map-based Monte Carlo (MMC) simulation, to stochastically extract folding pathways from the map. We demonstrate that our technique can be applied to large RNA (e.g., 200+ nucleotides), where representing the full landscape is infeasible, and that our tools provide results comparable to other simulation methods that work on complete energy landscapes. We present results showing that our approach computes the same relative functional rates as seen in experiments for the relative plasmid replication rates of ColE1 RNAII and its mutants, and for the relative gene expression rates of MS2 phage RNA and its mutants.
Parallel Protein Folding with STAPL, Shawna Thomas, Gabriel Tanase, Lucia K. Dale, Jose E. Moreira, Lawrence Rauchwerger, Nancy M. Amato, Concurrency and Computation: Practice and Experience, Vol: 17, Issue: 14, pp. 1643-1656, Dec 2005. DOI: https://doi.org/10.1002/cpe.950
Keywords: Parallel Planning, Protein Folding, STAPL
Links : [Published]
BibTex
@article{https://doi.org/10.1002/cpe.950,
author = {Thomas, Shawna and Tanase, Gabriel and Dale, Lucia K. and Moreira, Jose M. and Rauchwerger, Lawrence and Amato, Nancy M.},
title = {Parallel protein folding with STAPL},
journal = {Concurrency and Computation: Practice and Experience},
volume = {17},
number = {14},
pages = {1643-1656},
keywords = {protein folding, motion planning, parallel libraries, C++},
doi = {https://doi.org/10.1002/cpe.950},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/cpe.950},
eprint = {https://onlinelibrary.wiley.com/doi/pdf/10.1002/cpe.950},
abstract = {Abstract The protein-folding problem is a study of how a protein dynamically folds to its so-called native state—an energetically stable, three-dimensional conformation. Understanding this process is of great practical importance since some devastating diseases such as Alzheimer's and bovine spongiform encephalopathy (Mad Cow) are associated with the misfolding of proteins. We have developed a new computational technique for studying protein folding that is based on probabilistic roadmap methods for motion planning. Our technique yields an approximate map of a protein's potential energy landscape that contains thousands of feasible folding pathways. We have validated our method against known experimental results. Other simulation techniques, such as molecular dynamics or Monte Carlo methods, require many orders of magnitude more time to produce a single, partial trajectory. In this paper we report on our experiences parallelizing our method using STAPL (Standard Template Adaptive Parallel Library) that is being developed in the Parasol Lab at Texas A\&M. An efficient parallel version will enable us to study larger proteins with increased accuracy. We demonstrate how STAPL enables portable efficiency across multiple platforms, ranging from small Linux clusters to massively parallel machines such as IBM's BlueGene/L, without user code modification. Copyright © 2005 John Wiley \& Sons, Ltd.},
year = {2005}
}
Abstract
The protein-folding problem is a study of how a protein dynamically folds to its so-called native state - an energetically stable, three-dimensional conformation. Understanding this process is of great practical importance since some devastating diseases such as Alzheimer's and bovine spongiform encephalopathy (Mad Cow) are associated with the misfolding of proteins. We have developed a new computational technique for studying protein folding that is based on probabilistic roadmap methods for motion planning. Our technique yields an approximate map of a protein's potential energy landscape that contains thousands of feasible folding pathways. We have validated our method against known experimental results. Other simulation techniques, such as molecular dynamics or Monte Carlo methods, require many orders of magnitude more time to produce a single, partial trajectory. In this paper we report on our experiences parallelizing our method using STAPL (Standard Template Adaptive Parallel Library) that is being developed in the Parasol Lab at Texas A&M. An efficient parallel version will enable us to study larger proteins with increased accuracy. We demonstrate how STAPL enables portable efficiency across multiple platforms, ranging from small Linux clusters to massively parallel machines such as IBM's BlueGene/L, without user code modification.
Protein Folding by Motion Planning, Shawna Thomas, Guang Song, Nancy M. Amato, Physical Biology, Vol: 2, Issue: 4, pp. S148-S155, Nov 2005. DOI: 10.1088/1478-3975/2/4/S09
Keywords: Protein Folding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@article{Thomas_2005,
doi = {10.1088/1478-3975/2/4/s09},
url = {https://doi.org/10.1088%2F1478-3975%2F2%2F4%2Fs09},
year = 2005,
month = {nov},
publisher = {{IOP} Publishing},
volume = {2},
number = {4},
pages = {S148--S155},
author = {Shawna Thomas and Guang Song and Nancy M Amato},
title = {Protein folding by motion planning},
journal = {Physical Biology},
abstract = {We investigate a novel approach for studying protein folding that has evolved from robotics motion planning techniques called probabilistic roadmap methods (PRMs). Our focus is to study issues related to the folding process, such as the formation of secondary and tertiary structures, assuming we know the native fold. A feature of our PRM-based framework is that the large sets of folding pathways in the roadmaps it produces, in just a few hours on a desktop PC, provide global information about the protein\'s energy landscape. This is an advantage over other simulation methods such as molecular dynamics or Monte Carlo methods which require more computation and produce only a single trajectory in each run. In our initial studies, we obtained encouraging results for several small proteins. In this paper, we investigate more sophisticated techniques for analyzing the folding pathways in our roadmaps. In addition to more formally revalidating our previous results, we present a case study showing that our technique captures known folding differences between the structurally similar proteins G and L.}
}
Abstract
We investigate a novel approach for studying protein folding that has evolved from robotics motion planning techniques called probabilistic roadmap methods (PRMs). Our focus is to study issues related to the folding process, such as the formation of secondary and tertiary structures, assuming we know the native fold. A feature of our PRM-based framework is that the large sets of folding pathways in the roadmaps it produces, in just a few hours on a desktop PC, provide global information about the protein\'s energy landscape. This is an advantage over other simulation methods such as molecular dynamics or Monte Carlo methods which require more computation and produce only a single trajectory in each run. In our initial studies, we obtained encouraging results for several small proteins. In this paper, we investigate more sophisticated techniques for analyzing the folding pathways in our roadmaps. In addition to more formally revalidating our previous results, we present a case study showing that our technique captures known folding differences between the structurally similar proteins G and L.
Iterative Relaxation of Constraints: A Framework for Improving Automated Motion Planning, O. Burchan Bayazit, Dawen Xie, Nancy M. Amato, In Proc. IEEE Int. Conf. Intel. Rob. Syst. (IROS), pp. 586-593, Edmonton, Alberta, Canada, Aug 2005. DOI: 10.1109/IROS.2005.1545045
Keywords: Computational Biology, Sampling-Based Motion Planning, Virtual Reality
Links : [Published]
BibTex
@INPROCEEDINGS{1545045,
author={O. B. {Bayazit} and {Dawen Xie} and N. M. {Amato}},
booktitle={2005 IEEE/RSJ International Conference on Intelligent Robots and Systems}, title={Iterative relaxation of constraints: a framework for improving automated motion planning},
year={2005},
volume={},
number={},
pages={3433-3440},
doi={10.1109/IROS.2005.1545045}}
Abstract
This paper presents a technique for improving the efficiency of automated motion planners. Motion planning has application in many areas such as robotics, virtual reality systems, computer-aided design, and even computational biology. Although there have been steady advances in motion planning algorithms, especially in randomized approaches such as probabilistic roadmap methods (PRMs) or rapidly-exploring random trees (RRTs), there are still some classes of problems that cannot be solved efficiently using these state-of-the-art motion planners. In this paper, we suggest an iterative strategy addressing this problem where we first simplify the problem by relaxing some feasibility constraints, solve the easier version of the problem, and then use that solution to help us find a solution for the harder problem. We show how this strategy can be applied to rigid bodies and to linkages with high degrees of freedom, including both open and closed chain systems. Experimental results are presented for linkages composed of 9-98 links. Although we use PRMs as the automated planner, the framework is general and can be applied with other motion planning techniques as well.
C-Space Subdivision and Integration in Feature-Sensitive Motion Planning, Marco A. Morales A., Lydia Tapia, Roger Pearce, Samuel Rodriguez, Nancy M. Amato, In Proc. IEEE International Conference on Robotics and Automation (ICRA), pp. 3114-3119, Barcelona, Spain, Apr 2005. DOI: 10.1109/ROBOT.2005.1570589
Keywords: Machine Learning, Motion Planning, Sampling-Based Motion Planning
Links : [Published] [Manuscript]
BibTex
@INPROCEEDINGS{1570589,
author={M. A. {Morales A.} and L. {Tapia} and R. {Pearce} and S. {Rodriguez} and N. M. {Amato}},
booktitle={Proceedings of the 2005 IEEE International Conference on Robotics and Automation},
title={C-space Subdivision and Integration in Feature-Sensitive Motion Planning},
year={2005},
volume={},
number={},
pages={3114-3119},
doi={10.1109/ROBOT.2005.1570589}}
Abstract
There are many randomized motion planning techniques, but it is often difficult to determine what planning method to apply to best solve a problem. Planners have their own strengths and weaknesses, and each one is best suited to a specific type of problem. In previous work, we proposed a meta-planner that, through analysis of the problem features, subdivides the instance into regions and determines which planner to apply in each region. The results obtained with our prototype system were very promising even though it utilized simplistic strategies for all components. Even so, we did determine that strategies for problem subdivision and for combination of partial regional solutions have a crucial impact on performance. In this paper, we propose new methods for these steps to improve the performance of the meta-planner. For problem subdivision, we propose two new methods: a method based on ÃÂÃÂÃÂâÃÂÃÂÃÂÃÂÃÂÃÂÃÂàgapsÃÂÃÂÃÂâÃÂÃÂÃÂÃÂÃÂÃÂÃÂàand a method based on information theory. For combining partial solutions, we propose two new methods that concentrate on neighboring areas of the regional solutions. We present results that show the performance gain achieved by utilizing these new strategies.
Using Motion Planning to Map Protein Folding Landscapes and Analyze Folding Kinetics of Known Native Structures , Nancy M. Amato, Ken A. Dill, Guang Song, Journal of Computational Biology, Vol: 10, Issue: 3-4, pp. 239-255, Jul 2004. DOI: 10.1089/10665270360688002
Keywords: Computational Biology, Protein Folding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@article{10.1089/10665270360688002,
author ={Nancy M. {Amato}, Ken {Dill}, Guang {Song}},
title ={Using Motion Planning to Map Protein Folding Landscapes and Analyze Folding Kinetics of Known Native Structures},
journal ={Journal of Computational Biology},
volume ={10},year ={2004},number ={3-4},pages ={239-255},
doi={10.1089/10665270360688002}
}
Abstract
We investigate a novel approach for studying the kinetics of protein folding. Our framework has evolved from robotics motion planning techniques called probabilistic roadmap methods (PRMs) that have been applied in many diverse fields with great success. In our previous work, we presented our PRM-based technique and obtained encouraging results studying protein folding pathways for several small proteins. In this paper, we describe how our motion planning framework can be used to study protein folding kinetics. In particular, we present a refined version of our PRM-based framework and describe how it can be used to produce potential energy landscapes, free energy landscapes, and many folding pathways all from a single roadmap which is computed in a few hours on a desktop PC. Results are presented for 14 proteins. Our ability to produce large sets of unrelated folding pathways may potentially provide crucial insight into some aspects of folding kinetics, such as proteins that exhibit both two-state and three-state kinetics that are not captured by other theoretical techniques.
Parallel Protein Folding with STAPL, Shawna Thomas, Nancy M. Amato, 18th International Parallel and Distributed Processing Symposium (IPDPS), pp. 189-, Santa Fe, NM, USA, Apr 2004. DOI: 10.1109/IPDPS.2004.1303204
Keywords: Parallel Planning, Protein Folding, STAPL
Links : [Published]
BibTex
@INPROCEEDINGS{1303204,
author={S. {Thomas} and N. M. {Amato}},
booktitle={18th International Parallel and Distributed Processing Symposium, 2004. Proceedings.},
title={Parallel protein folding with STAPL},
year={2004},
volume={},
number={},
pages={189-},
doi={10.1109/IPDPS.2004.1303204}}
Abstract
Summary form only given. The protein folding problem is to study how a protein dynamically folds to its so-called native state - an energetically stable, three-dimensional configuration. Understanding this process is of great practical importance since some devastating diseases such as Alzheimer\'s and bovine spongiform encephalopathy (Mad Cow) are associated with the misfolding of proteins. In our group, we have developed a new computational technique for studying protein folding that is based on probabilistic roadmap methods for motion planning. Our technique yields an approximate map of a protein\'s potential energy landscape that contains thousands of feasible folding pathways. We have validated our method against known experimental results. Other simulation techniques, such as molecular dynamics or Monte Carlo methods, require many orders of magnitude more time to produce a single, partial, trajectory. We report on our experiences parallelizing our method using STAPL (the standard template adaptive parallel library), that is being developed in the Parasol Lab at Texas A&M. An efficient parallel version enables us to study larger proteins with increased accuracy. We demonstrate how STAPL enables portable efficiency across multiple platforms without user code modification. We show performance gains on two systems: a dedicated Linux cluster and an extremely heterogeneous multiuser Linux cluster.
Using Motion Planning to Study RNA Folding Kinetics, Xinyu Tang, Bonnie Kirkpatrick, Shawna Thomas, Guang Song, Nancy M. Amato, In Proc. Int. Conf. Comput. Molecular Biology (RECOMB), pp. 252-261, Mar 2004. DOI: 10.1145/974614.974648
Keywords: Computational Biology, RNA, Sampling-Based Motion Planning
Links : [Published]
BibTex
@inproceedings{10.1145/974614.974648,
author = {Tang, Xinyu and Kirkpatrick, Bonnie and Thomas, Shawna and Song, Guang and Amato, Nancy M.},
title = {Using Motion Planning to Study RNA Folding Kinetics},
year = {2004},
isbn = {1581137559},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/974614.974648},
doi = {10.1145/974614.974648},
abstract = {We propose a novel, motion planning based approach to approximately map the energy landscape of an RNA molecule. Our method is based on the successful probabilistic roadmap motion planners that we have previously successfully applied to protein folding. The key advantage of our method is that it provides a sparse map that captures the main features of the landscape and which can be analyzed to compute folding kinetics. In this paper, we provide evidence that this approach is also well suited to RNA. We compute population kinetics and transition rates on our roadmaps using the master equation for a few moderately sized RNA and show that our results compare favorably with results of other existing methods.},
booktitle = {Proceedings of the Eighth Annual International Conference on Resaerch in Computational Molecular Biology},
pages = {252–261},
numpages = {10},
keywords = {motion planning, RNA, folding kinetics},
location = {San Diego, California, USA},
series = {RECOMB \'04}
}
Abstract
We propose a novel, motion planning based approach to approximately map the energy landscape of an RNA molecule. Our method is based on the successful probabilistic roadmap motion planners that we have previously successfully applied to protein folding. The key advantage of our method is that it provides a sparse map that captures the main features of the landscape and which can be analyzed to compute folding kinetics. In this paper, we provide evidence that this approach is also well suited to RNA. We compute population kinetics and transition rates on our roadmaps using the master equation for a few moderately sized RNA and show that our results compare favorably with results of other existing methods.
Neuron PRM: A Framework for Constructing Cortical Networks, Jyh-Ming Lien, Marco Morales, Nancy M. Amato, Neurocomputing, Vol: 54, pp. 191-197, Jun 2003. DOI: 10.1016/S0925-2312(02)00728-2
Keywords: Machine Learning, Sampling-Based Motion Planning
Links : [Published]
BibTex
@article{LIEN2003191,
title = "Neuron PRM: a framework for constructing cortical networks",
journal = "Neurocomputing",
volume = "52-54",
pages = "191 - 197",
year = "2003",
note = "Computational Neuroscience: Trends in Research 2003",
issn = "0925-2312",
doi = "https://doi.org/10.1016/S0925-2312(02)00728-2",
url = "http://www.sciencedirect.com/science/article/pii/S0925231202007282",
author = "Jyh-Ming Lien and Marco Morales and Nancy M. Amato",
keywords = "Cortical networks, PRM, BTS, L-system, Rectangle tree",
abstract = "The brain's extraordinary computational power to represent and interpret complex natural environments is essentially determined by the topology and geometry of the brain's architectures. We present a framework to construct cortical networks which borrows from probabilistic roadmap methods developed for robotic motion planning. We abstract the network as a large-scale directed graph, and use L-systems and statistical data to ‘grow’ neurons that are morphologically indistinguishable from real neurons. We detect connections (synapses) between neurons using geometric proximity tests."
}
Abstract
The brain's extraordinary computational power to represent and interpret complex natural environments is essentially determined by the topology and geometry of the brain's architectures. We present a framework to construct cortical networks which borrows from probabilistic roadmap methods developed for robotic motion planning. We abstract the network as a large-scale directed graph, and use L-systems and statistical data to âÃÂÃÂgrowâÃÂàneurons that are morphologically indistinguishable from real neurons. We detect connections (synapses) between neurons using geometric proximity tests.
A Path Planning-based Study of Protein Folding With a Case Study of Hairpin Formation in Protein G and L, Guang Song, Shawna Thomas, Ken A. Dill, J. Martin Scholtz, Nancy M. Amato, In Proc. Pac. Symp. of Biocomputing (PSB), Vol: 8, pp. 240-251, Lihue, Hawaii, Jan 2003.
Keywords: Computational Biology, Protein Folding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@INPROCEEDINGS{Song_2003,
author={Guang {Song}, Shawna {Thomas}, Ken A. {Dill}, J. Martin {Scholtz}, Nancy M. {Amato}},
booktitle={In Proc. Pac. Symp. of Biocomputing (PSB)},
title={A Path Planning-based Study of Protein Folding With a Case Study of Hairpin Formation in Protein G and L},
year={2003}, volume={8}, number={}, pages={240-251},
doi={}
}
Abstract
We investigate a novel approach for studying protein folding that has evolvedfrom robotics motion planning techniques calledprobabilistic roadmapmethods(prms). Our focus is to study issues related to the folding process, such as theformation of secondary and tertiary structure,assumingweknowthenativefold.A feature of ourprm-based framework is that the large sets of folding pathwaysin the roadmaps it produces, in a few hours on a desktop PC, provide globalinformation about the proteinÃÂâÃÂÃÂÃÂÃÂs energy landscape. This is an advantage over othersimulation methods such as molecular dynamics or Monte Carlo methods whichrequire more computation and produce only a single trajectory in each run. Inour initial studies, we obtained encouraging results for several small proteins. Inthis paper, we investigate more sophisticated techniques for analyzing the foldingpathways in our roadmaps. In addition to more formally revalidating our previousresults, we present a case study showing our technique captures known foldingdifferences between the structurally similar proteins G and L.
A motion planning approach to folding: from paper craft to protein folding, Guang Song, Nancy M. Amato, In Proc. IEEE Int. Conf. Robot. Autom. (ICRA), Vol: 1, pp. 948-953, Seoul, Korea, May 2001. DOI: 10.1109/ROBOT.2001.932672
Keywords: Path Planning, Protein Folding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@INPROCEEDINGS{932672,
author={ {Guang Song} and N. M. {Amato}},
booktitle={Proceedings 2001 ICRA. IEEE International Conference on Robotics and Automation (Cat. No.01CH37164)}, title={A motion planning approach to folding: from paper craft to protein folding},
year={2001},
volume={1},
number={},
pages={948-953 vol.1},
doi={10.1109/ROBOT.2001.932672}}
Abstract
We present a framework for studying folding problems from a motion planning perspective. Modeling foldable objects as tree-like multi-link objects allows one to apply motion planning techniques to folding problems. An important feature of this approach is that it not only allows one to study foldability questions, such as, can an object be folded (or unfolded) into another object, but also provides one with another tool for investigating the dynamic folding process itself. The framework proposed here has application to traditional motion planning areas such as automation and animation, and presents a novel approach for studying protein folding pathways. Preliminary experimental results with traditional paper crafts (e.g., box folding) and small proteins (approximately 60 residues) are quite encouraging.
Ligand Binding with OBPRM and Haptic User Input: Enhancing Automatic Motion Planning with Virtual Touch, O. Burchan Bayazit , Guang Song , Nancy M. Amato , ACM Digital Library, College Station, Texas, USA, Oct 2000.
Keywords: Ligand Binding, Sampling-Based Motion Planning
Links : [Published]
BibTex
@MISC{Bayazit00ligandbinding,
author = {O. Burchan Bayazit and Guang Song and Nancy M. Amato},
title = {Ligand Binding with OBPRM and Haptic User Input: Enhancing Automatic Motion Planning with Virtual Touch},
year = {2000}
}
Abstract
In this paper, we present a framework for studying ligand binding which is based on techniques recently developed in the robotics motion planning community. We are especially interested in locating binding sites on the protein for a ligand molecule. Our work investigates the performance of a fully automated motion planner, as well improvements obtained when supplementary user input is collected using a haptic device. Our results applying an obstacle-based probabilistic roadmap motion planning algorithm (obprm) to some known protein-ligand pairs are very encouraging. In particular, we were able to automatically generate congurations close to, and correctly identify, the true binding site in the three protein-ligand complexes we tested. We nd that user input helps the planner, and a haptic device helps the user to understand the protein structure by enabling them to feel the forces which are hard to visualize.